
Etablierung und Optimierung neuer Methoden
Für massenspektrometrie-basierte Proteomanalysen setzen wir hauptsächlich auf drei technische Bereiche, die die moderne, identifikationsorientierte und quantitative Proteomics repräsentieren: Shot-gun Proteomanalysen / datenabhängige Massenspektrometrie, SRM-basierte / zielgerichtete Massenspektrometrie und datenunabhängigen Massenspektrometrie. Für das Erreichen der maximalen Leistung sind wir stark vom technologischen Fortschritt der massenspektrometrischen Ausstattung abhängig. Um die diese moderne Ausstattung in angemessener Weise zu nutzen müssen Arbeitsabläufe optimiert oder neu entwickelt werden. Diese Bemühungen sind vor allem deshalb bedeutsam, weil das experimentelle Vorgehen häufig mehr über die wissenschaftliche Erkenntnisse entscheidet als die Geräte selbst.
- Becher et al. PMID: 23894095.
FISH-FACS-Proteomics für die Anreicherung nicht-kultivierbarer Mikroorganismen aus Umweltproben
Mikrobielle Gemeinschaften werden umfassend untersucht, um ihre Rolle in der Umwelt zu verstehen. Um ein tieferes Verständnis für ihre Wechselwirkungen, funktionellen Rollen und ihrer Anpassungsfähigkeit an die Umwelt zu gewinnen, sind jedoch detaillierte Einblicke in die wichtigsten mikrobiellen Kladen innerhalb dieser Gemeinschaften notwendig. Mit Metaproteomics können mikrobiellen Stoffwechselaktivitäten in vielfältigen Umgebungen untersucht werden. Die Funktionsanalyse spezifischer Mikroorganismen wird jedoch aufgrund des Problems der Proteininferenz, das durch Sequenzhomologien zwischen eng verwandten Arten entsteht, erschwert. Diese Herausforderung schränkt unser Verständnis der Rolle bestimmter Mikroben in komplexen Umweltproben ein. Wir haben eine Methode entwickelt, die Fluoreszenz-in-situ-Hybridisierung (FISH) und fluoreszenzaktivierte Zellsortierung (FACS) mit massenspektrometriebasierter Proteomik kombiniert, um Proteine von bisher nicht kultivierbaren Bakterien direkt aus Umweltproben zu analysieren. Tatsächlich reichen inzwischen Proben mit nur 1 × 10⁵ bakteriellen Zellen für eine zuverlässige qualitative Proteinidentifizierung aus, während Proben mit 5 × 10⁵ bis 1 × 10⁶ Zellen eine reproduzierbare Proteinquantifizierung ermöglichen. Darüber hinaus verbessert die Verwendung einer taxonspezifischen Datenbank die Datenanalyse, indem sie die Größe der Proteingruppen im Vergleich zu Metaproteomikdaten deutlich reduziert.
- Kale et al. PMID: 40989907
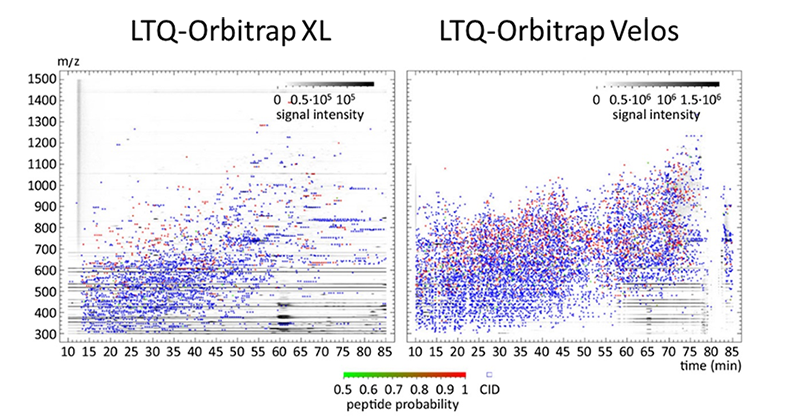
Data-independent Acquistion für die robuste und sensitive Identifikation von Proteinen
Obwohl die datenabhängige Erfassung von MS-Spektren (DDA) nach wie vor der am häufigsten verwendete Ansatz in Standard-Proteomik-Workflows ist, haben wir für viele unserer Analysen eine datenunabhängige Erfassungsstrategie (DIA) implementiert. Im Gegensatz zur DDA werden bei der DIA alle Vorläuferionen innerhalb vordefinierter Masse-zu-Ladungs-Fenster (m/z) unabhängig von ihrer Intensität systematisch fragmentiert. Durch den Einsatz von DIA verbessern wir die Identifikationsrate von gering abundanten Peptiden, die bei DDA möglicherweise übersehen würden, was auch in einer reduzierten Anzahl von missing values in quantitativen Datensätze resultiert.
- Bruderer et al. PMID: 29070702
- Zhang et al. PMID: 32275110
- Wu et al. PMID: 39152734
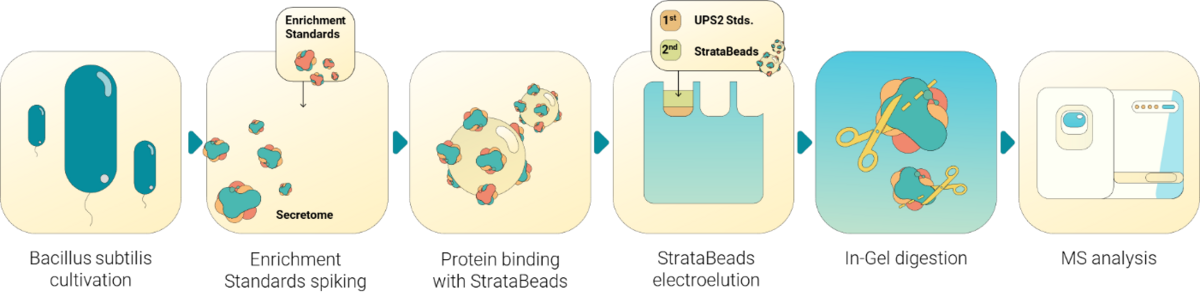
Absolute Quantifizierung von cytosolischen Proteinen, Membranproteinen und extrazellulären Proteinen
Die absolute Quantifizierung, die vor allem in systembiologischen Studien verfolgt wird, stellt eine anspruchsvolle technische und biologische Fragestellung in der Proteomics dar. Für die umfassendsten und genauesten Datensätze werden absolute Proteinmengen benötigt, die in Kombination mit anderen „Omics“-Daten einen tiefen modellhaften Einblick in die intrazellulär regulierten Prozesse ermöglichen. Inzwischen haben wir Methoden zur absoluten Quantifizierung von Proteinen verschiedener Lokalisationen entwickelt. Damit können wir nun für mehrere tausend Proteine aus dem Cytosol, der bakteriellen Membran und dem Kulturüberstand die exakte Menge bestimmen. Hierfür verwenden wir geeignete Standards, die nicht nur erlauben, relative Proteinmengen in absolute Werte umzurechnen, sondern auch die unterschiedlichen Anreicherungsschritten, z.B. bei der Gewinnung von Membranproteinen oder sekretierten Proteinen, korrekt berücksichtigen. Da die umfangreiche Auswertung der Messergebnisse ein mehrstufiges Transformieren der MS-Rohdaten erfordert, kann der Prozess vor allem für nicht spezialisierte Anwender fehleranfällig sein. Wir haben deshalb Alpaca Proteomics entwickelt, ein anwenderfreundliches Tool zur Prozessierung von MS-Daten für die Generierung von absoluten Messdaten. (Link)
- Maass et al. PMID: 21395229.
- Muntel et al. PMID: 24696501.
- Antelo-Varela et al. PMID: 31424929.
- Ferrero-Bordera et al. PMID: 38358275.
- Ferrero-Bordera et al. PMID: 40285550.
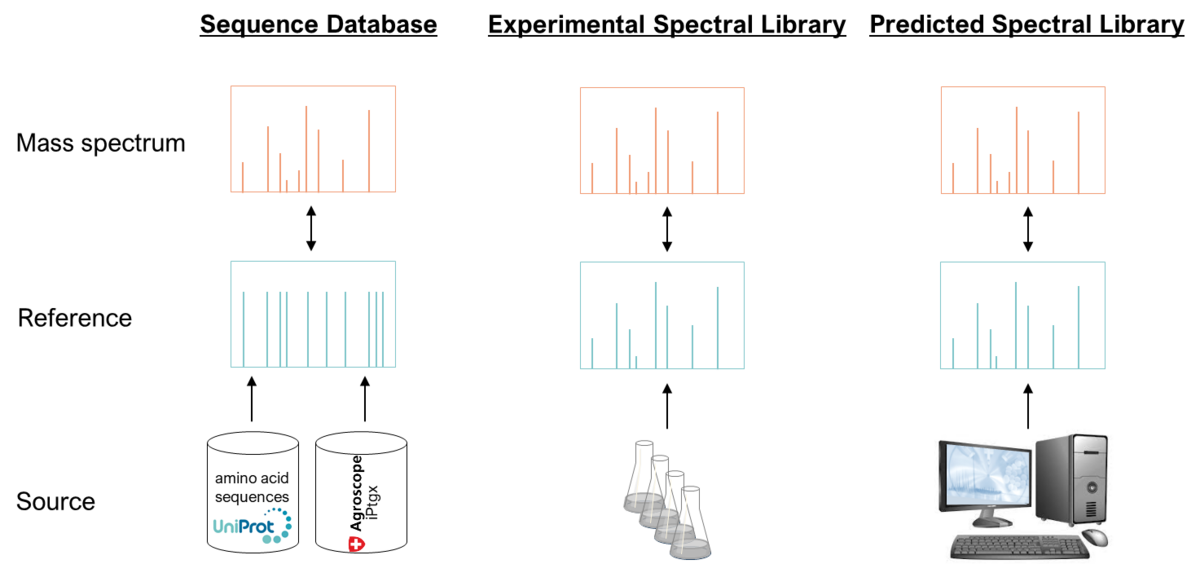
Identifikation und Quantifizierung (kleiner) Proteine mit Spektrenbibliotheken
Die analytischen Herausforderungen bei der Detektion kleiner Proteine. führten dazu, dass diese in aktuellen proteomischen Studien unterrepräsentiert sind, obwohl gezeigt wurde, dass diese Proteine häufig an wichtigen physiologischen Funktionen beteiligt sind. Wir haben daher verschiedene Methoden der Datenprozessierung systematisch evaluiert und konnten zeigen, dass die Detektion und Quantifizierung kleiner Proteine von der Anwendung experimenteller Spektrenbibliotheken profitiert.
- Bartel et al. PMID: 41613483.
Zielgerichtete Analyse von einzelnen Proteinen
In verschiedenen spezialisierten Fällen kann es sinnvoll sein, ausgewählte Proteine und Peptide mit zielgerichteten MS-Ansätzen wie (selected reaction monitoring, SRM, oder parallel reaction monitoring, PRM) zu untersuchen. Mögliche Applikation umfassen die absolute Proteinquantifizierung, Validierung von nicht-annotierten Proteinen, Untersuchungen zur Lokalisation von post-translationalen Modifikationen und die sensitive Detektion von gering abundanten Proteinen in komplexen Proben.
- Maass et al. PMID: 21395229.
- Hentschker et al. PMID: 32154730.
- Hadjeras et al. PMID: 37223747.
- Bartel et al. PMID: 32812434.
Proteomweite Analyse posttranslationaler Modifikationen
Alle Organismen sind durch eine hohe Dynamik in der Proteinbiosynthese und ihrer Abbauprozesse charakterisiert. Durch zelluläre Herausforderungen und Anpassungsreaktionen verursachte posttranslationale Modifikationsereignisse führen zu einer einzigartigen Regulation der Aktivität der in der Zelle vorhandenen Proteine. Dazu gehören neben Phosphorylierungen und Acetylierungen, auch Proteinoxidation oder Ubiquitinierungen. Wir arbeiten daraufhin, die molekularen Ziele dieser Modifikationen unter Verwendung von MS- basierten Proteomanalysen zu bestimmen. Dabei sind wir stetig bestrebt, sowohl die Probenvorbereitung, die MS-basierte Probenanalyse als auch die Datenprozessierung an die Analyse und Charakterisierung dieser herausfordernden (Sub)Proteome anzupassen.
- Walgraeve et al. PMID: 37819171.
- Sura et al. PMID: 35281454.
- Hentschker et al. PMID: 32154730.
- Junker et al. PMID: 30358407.