
Establishment and optimization of new methods
For mass spectrometry based proteomics we rely mainly on three technical fields that represent the modern, discovery driven, quantitative proteomics : Shot-gun proteomics/ data dependent mass spectrometry, SRM-based/ targeted mass spectrometry and data independent mass spectrometry. To achieve maximum performance we are strongly dependent on technological advances by mass spectrometric equipment. But this high-end-equipment must be also exploited in a proper way, workflows have to be optimized or newly developed. These efforts are not less important as frequently protocols decide over scientific output rather instrumentation.
- Becher et al. PMID: 23894095.
FISH–FACS proteomics for the enrichment of non-cultivable microorganisms from environmental samples
Microbial communities are extensively studied to understand their roles in the environment. However, gaining deeper insight into their interactions, functional roles, and environmental adaptability requires detailed characterization of key microbial clades within these communities. Metaproteomics enables the investigation of microbial metabolic activities across diverse environments, but functional analysis of specific microorganisms is hindered by the protein inference problem, which arises from sequence homology among closely related species. This limitation constrains our ability to assign functions to individual taxa in complex environmental samples.
To address this challenge, we have developed a method that combines fluorescence in situ hybridization (FISH) and fluorescence-activated cell sorting (FACS) with mass spectrometry–based proteomics, enabling the direct analysis of proteins from previously non-cultivable bacteria in environmental samples. Notably, samples containing as few as 1 × 10⁵ bacterial cells are sufficient for reliable qualitative protein identification, while 5 × 10⁵ to 1 × 10⁶ cells enable reproducible protein quantification. Furthermore, the use of a taxon-specific database improves data analysis by substantially reducing protein group complexity compared to conventional metaproteomic datasets.
- Kale et al. PMID: 40989907
Data-independent acquisition for robust and sensitive protein identification
Although data-dependent acquisition (DDA) of MS spectra remains the most widely used approach in standard proteomics workflows, we have implemented a data-independent acquisition (DIA) strategy for many of our analyses. In contrast to DDA, DIA systematically fragments all precursor ions within predefined mass-to-charge (m/z) windows, independent of their intensity. By applying DIA, we improve the identification of low-abundance peptides that may be missed in DDA approaches, which also results in a reduced number of missing values in quantitative datasets.
- Bruderer et al. PMID: 29070702
- Zhang et al. PMID: 32275110
- Wu et al. PMID: 39152734
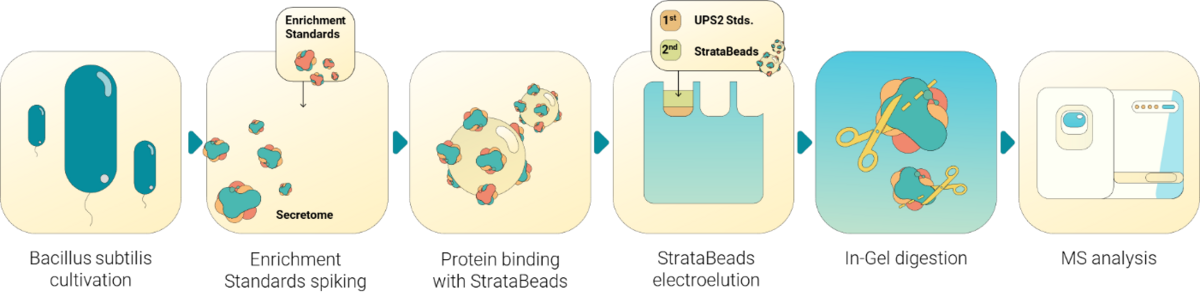
Absolute quantification of cytosolic, membrane, and extracellular proteins
Absolute quantification, particularly pursued in systems biology studies, represents a technically and biologically demanding challenge in proteomics. Comprehensive and accurate datasets require absolute protein abundances, which, when integrated with other omics data, enable a deeper, model-driven understanding of intracellular regulatory processes. We have developed methods for the absolute quantification of proteins across different cellular localizations. This allows us to determine the precise abundance of several thousand proteins from the cytosol, bacterial membranes, and culture supernatants. To achieve this, we employ suitable standards that not only enable the conversion of relative protein intensities into absolute values but also account for enrichment steps, such as those used for membrane or secreted protein fractions.
As the analysis of such datasets requires multi-step transformation of raw MS data, the process can be error-prone, particularly for non-specialized users. To address this, we developed Alpaca Proteomics, a user-friendly tool for processing MS data and generating absolute quantitative measurements. (Link)
- Maass et al. PMID: 21395229.
- Muntel et al. PMID: 24696501.
- Antelo-Varela et al. PMID: 31424929.
- Ferrero-Bordera et al. PMID: 38358275.
- Ferrero-Bordera et al. PMID: 40285550.
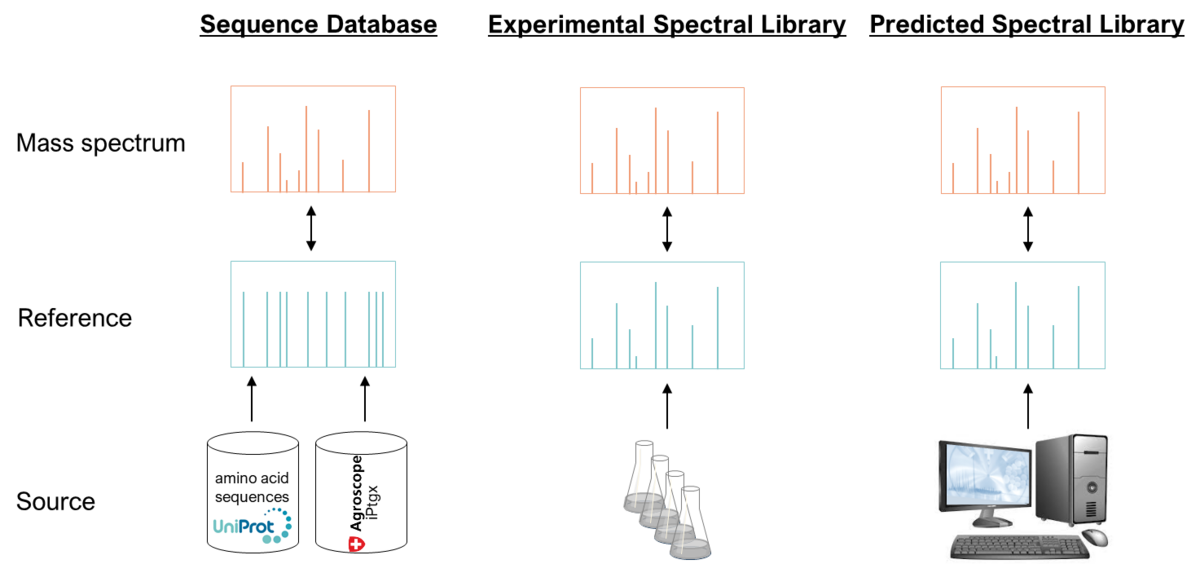
Identification and quantification of (small) proteins using spectral libraries
The analytical challenges associated with the detection of small proteins have led to their underrepresentation in current proteomics studies, despite evidence that they frequently play important physiological roles. We therefore systematically evaluated different data processing strategies and demonstrated that the detection and quantification of small proteins benefit from the use of experimental spectral libraries.
- Bartel et al. PMID: 41613483.
Implementation of targeted assays of single proteins
In specific applications, it can be advantageous to analyze selected proteins and peptides using targeted mass spectrometry approaches such as selected reaction monitoring (SRM) or parallel reaction monitoring (PRM). Typical applications include absolute protein quantification, validation of unannotated proteins, investigation of post-translational modification sites, and the sensitive detection of low-abundance proteins in complex samples.
- Maass et al. PMID: 21395229.
- Hentschker et al. PMID: 32154730.
- Hadjeras et al. PMID: 37223747.
- Bartel et al. PMID: 32812434.
Proteome-wide analysis of post-translational modifications
All organisms are characterized by highly dynamic processes of protein synthesis and degradation. Post-translational modification events, driven by cellular challenges and adaptive responses, result in a distinct regulation of protein activity within the cell. These include not only phosphorylation and acetylation, but also protein oxidation and ubiquitination. We aim to identify the molecular targets of these modifications using MS-based proteomics approaches. To this end, we continuously adapt sample preparation, mass spectrometric analysis, and data processing strategies to enable the analysis and characterization of these challenging (sub)proteomes.
- Walgraeve et al. PMID: 37819171.
- Sura et al. PMID: 35281454.
- Hentschker et al. PMID: 32154730.
- Junker et al. PMID: 30358407.