
Technical and methodological development for state-of-the-art proteomics
Sample processing is crucial for sensitive and comprehensive proteomic analyses, especially prior to mass spectrometric analysis in LC-MS/MS systems. Here we have developed and adapted a wide range of sophisticated protocols. Their elaborate use enables us to characterize proteomes of different microbes, both for our independent research and in cooperation with numerous national and international partners.
Some examples are:
Subcellular fractionation in bacteria
For a comprehensive and sensitive characterization of bacterial proteomes, fractionation of samples based on cellular and extracellular protein localization is often advantageous. Such fractionation enables targeted adaptation of extraction methods to the physicochemical properties of the proteins of interest and results in improved proteome coverage.
We have developed an efficient protocol for the enrichment of extremely dilute proteome samples, which can also be used for the recovery of extracellular proteins from growth media. Based on commercially available affinity beads, the method allows not only enrichment of low-abundance proteins but also sensitive sample purification. For example, salts or nucleic acids that would otherwise interfere with downstream mass spectrometric analyses can be effectively removed. A further notable feature of this protocol is the possibility to store and ship proteome samples without the need for complex cooling logistics.
- Bonn et al. PMID: 24987932.
- Otto et al. PMID: 28109424.
Although approximately one third of the genomes of most microorganisms encodes predicted membrane proteins, these proteins are often underrepresented in proteomics studies. To enable access to the membrane proteome, we have established and further developed protocols that selectively enrich proteins from the inner and outer membrane as well as membrane-associated proteins. These approaches enable sensitive LC–MS/MS analysis of different classes of membrane proteins.
- Dreisbach et al. PMID: 18491319.
- Wolff et al. PMID: 18460691.
- Maaß et al. PMID 30694509.
The bacterial surface also comprises numerous proteins with diverse physicochemical properties, including membrane proteins, lipoproteins, and proteins covalently or non-covalently attached to the cell wall. To specifically analyze these proteins, we have developed dedicated protocols, for example for the selective labeling of surface-exposed proteins by biotinylation or for the direct proteolytic digestion of surface proteins.
- Hempel et al. PMID: 20108986.
- Dreisbach et al. PMID: 21674804.
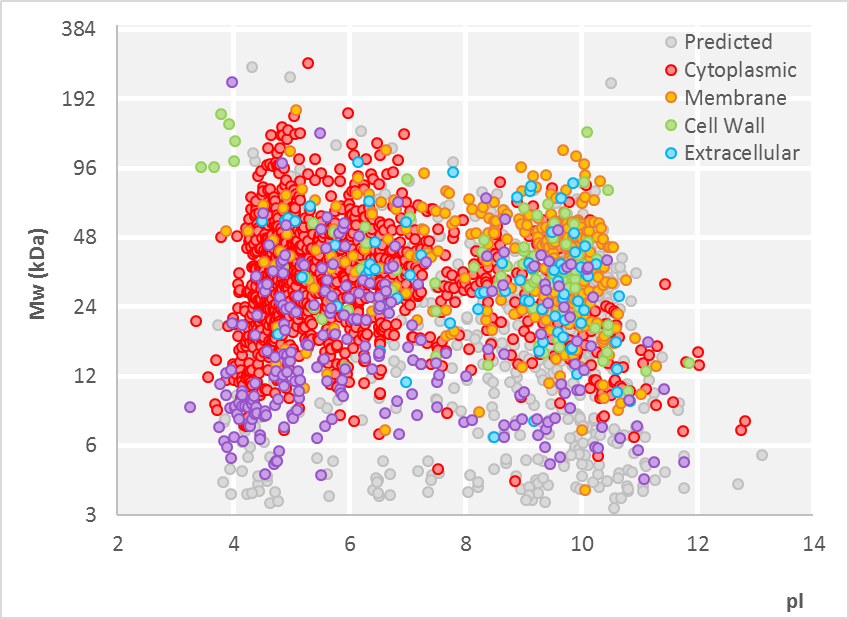
Enrichment of small proteins
The recent discovery of short open reading frames across all domains of life has made it necessary to validate their existence at the protein level and to assign functions to the encoded proteins. However, small proteins are analytically challenging. They are often lost during sample preparation and are frequently masked by larger proteins during ionization, separation, and detection in mass spectrometry. This hampers their efficient identification and quantitative characterization.
To improve access to the so-called “small proteome”, we have developed a method for the enrichment of small proteins based on solid-phase extraction, which was successfully applied to map the small proteome in Bacillus subtilis. Notably, the detection of small proteins strongly benefits from the use and combination of alternative proteases in addition to trypsin. Our tool CoMPaseD facilitates the identification of the most effective protease or protease combinations to achieve maximal proteome coverage, not limited to small proteins (Link).
- Bartel et al. PMID: 32812434.
- Cassidy et al. PMID: 34145981.
- Bartel et al. PMID: 41541604.
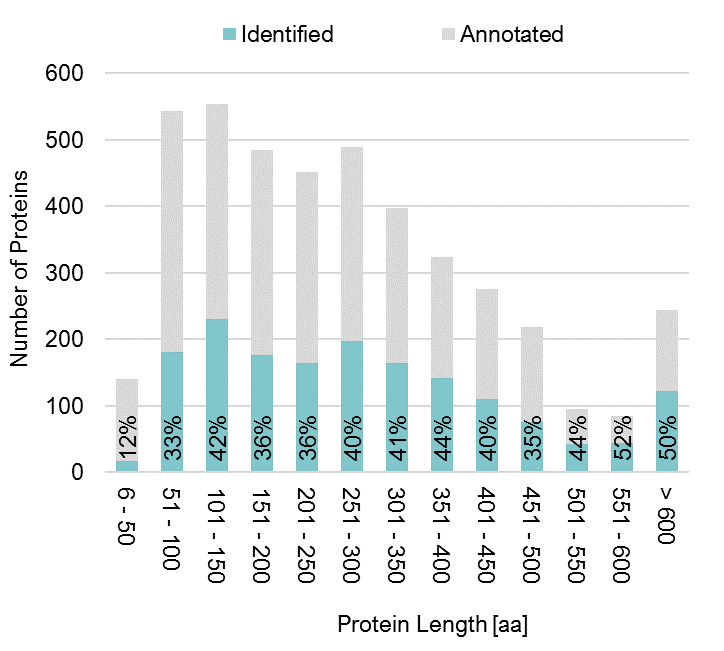
Extraction of proteins from clay-rich sediments
Another focus of our work lies in overcoming methodological barriers in the analysis of highly complex environmental samples. In particular, marine sediments represent a major challenge for metaproteomics due to their complex matrix, including high clay content and interfering humic substances. These components can bind proteins, rendering them inaccessible to mass spectrometric analysis and thereby hindering the identification of microbial functions within the sediments. To address these challenges, we have developed an optimized extraction workflow that combines the blocking of binding sites through the use of amino acids with highly efficient electroelution of proteins from sediment material.
Ostrizinski et al. PMID: 40568302.
Metaproteome analyses from seawater samples
A central focus of our research is the application and further development of metaproteomics to achieve a high-resolution understanding of the functional dynamics of microbial communities in complex marine ecosystems. Through the adaptation of an efficient protein extraction method, combined with highly sensitive mass spectrometry (LC–MS/MS), we are now able not only to characterize the composition of bacterioplankton communities but also to monitor their actual metabolic activities with near real-time resolution.
- Zühlke et al. PMID: 38757353
- Beidler et al. PMID: 38744821