
Establishment and optimization of new methods
For mass spectrometry based proteomics we rely mainly on three technical fields that represent the modern, discovery driven, quantitative proteomics : Shot-gun proteomics/ data dependent mass spectrometry, SRM-based/ targeted mass spectrometry and data independent mass spectrometry. To achieve maximum performance we are strongly dependent on technological advances by mass spectrometric equipment. But this high-end-equipment must be also exploited in a proper way, workflows have to be optimized or newly developed. These efforts are not less important as frequently protocols decide over scientific output rather instrumentation.
- Becher et al. PMID: 23894095.
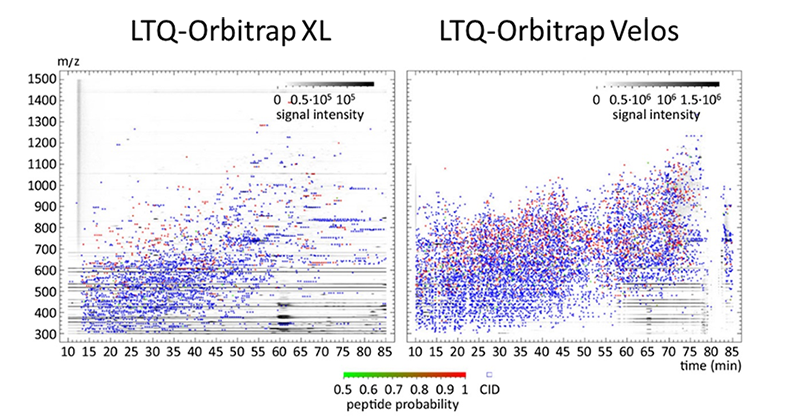
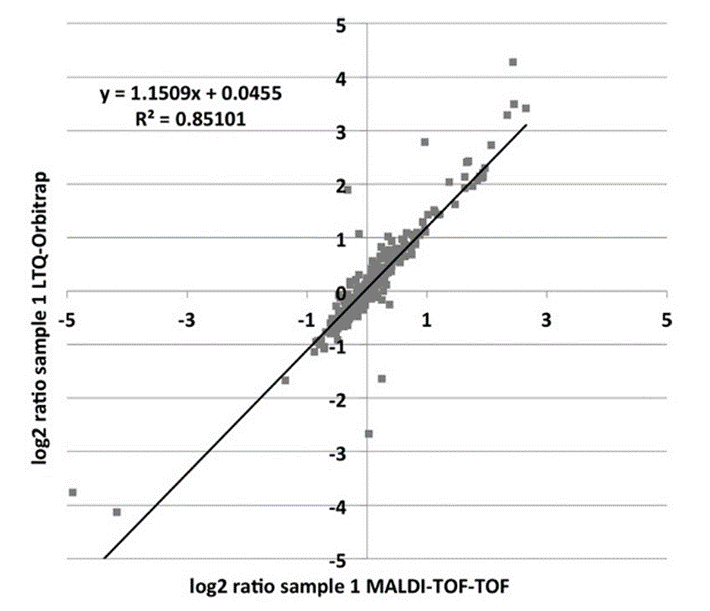
LC-MALDI
Despite the success of ESI- based mass spectrometry in the last years, MALDI mass spectrometry represents a reliable and fast technique for high throughput identification in classical proteomics. An advanced application is the spotting of eluting peptides from an LC run onto MALDI plates followed by MALDI-Tof/Tof identification / quantification. In such a way an entire LC run can be virtually frozen. This can be exploited for special analytic purposes, i.e. to find peptides of particular amino acid composition that are discriminated in processes of Electrospray ionization.
- Hessling et al. PMID: 23788530.
- Hessling et al. PMID: 24161710.
Large scale characterization of post- translational modifications
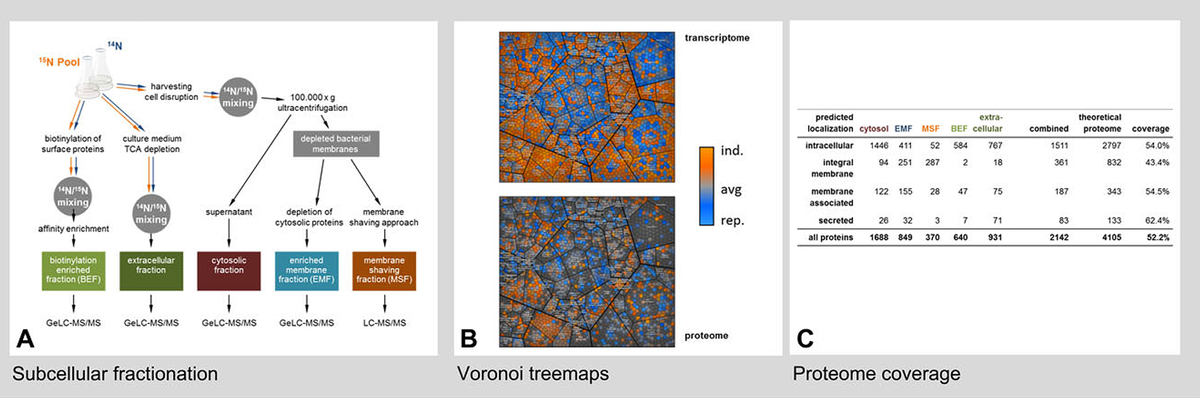
All organisms are characterized by highly dynamic protein synthesis and degradation processes. Additionally, post-translational modification events like phosphorylation or others, caused by different cellular challenges (e.g. oxidative stress damage/ protection), are leading to a peculiar regulation of the activities of existing proteins inside the cell. We aim at determining the molecular targets of these modifications both by very specific approaches employing Top- down proteomic techniques based on 2D gels and by large scale enrichment techniques (e.g. S/T/Y/R phosphorylations)
- Elsholz et al. PMID: 22517742.
- Bäsell et al. PMID: 24457182.
Relative quantification approaches – 15N labeling, SILAC
We have established a powerful workflow for quantitative microbial proteomics based on metabolic labeling with the heavy isotope of nitrogen (15N). A comparison of the same peptides that are composed of different isotopes serves as basis for determination of relative amounts. This technique can be easily employed for such microorganisms that are able to grow on defined (minimal) media. But also complex media are usable if they can be modified by exchange of all biological active nitrogen sources for their heavy counterparts. In our case we use both opportunities to feed different microorganisms and to generate heavy labeled proteome samples. Thus minimal media with heavy nitrogen salts ((NH4)2SO4 or (NH4)Cl) or complex media (based on labeled algae) can be used for relative quantitative proteomics.
- Otto et al. PMID: 24376008.
- Dreisbach et al. PMID: 18491319.
- Becher et al. PMID: 19997597.
- Otto et al. PMID: 21266987.
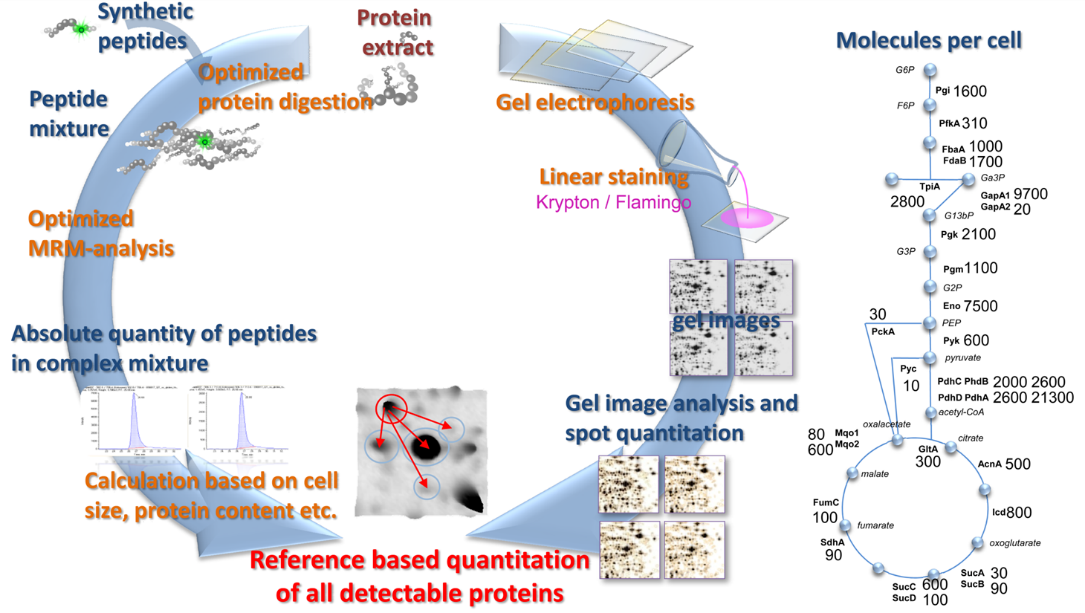
Absolute quantification approaches
Absolute quantification is one of the most challenging technical and biological tasks in proteomics that can be pursued especially in systems biology studies. Here, absolute data are required for most comprehensive and accurate datasets that can be modeled in combination with other “omics” data to achieve deeper insights into intracellular regulatory processes.
Examples for methods used in our lab are:
Implementation of targeted assays of single proteins (SRM)
For absolute quantification it is extremely crucial to compare endogenous peptides found in a proteome sample with artificially labeled, spiked peptides of known concentration. We employ this AQUA (Absolute quantification) approach regularly to determine single proteins, members of small protein complexes, or in combination with 2D Gel based proteomics on a global scale.
- Delumeau et al. PMID: 21710567.
- Maass et al. PMID: 21395229.
- Muntel et al. PMID: 24696501.
SRM-2D PAGE
For absolute quantification, we have combined two well-known and widely applied proteomic techniques, the separation of proteins via 2D gel electrophoresis and the absolute quantification of peptides by AQUA based on a Triple Quadrupole. Determination of absolute amounts of so- called “Anchor proteins” on 2D gels enables us to calibrate all other proteins found in the very same gel. It is the most cost effective method to determine the absolute protein concentration for several hundreds of proteins and their isoforms in complex proteomes to date.
- Maass et al. PMID: 21395229.
- Otto et al. PMID: 24376008.
MSE DIA
The second absolute quantification workflow is based on the MSE technology. Key feature is the data independent acquisition of precursor ions for MS/MS, and therefore MSE is perfectly suited for both an unbiased identification and quantification of complex proteomic samples. Absolute data are derived by a comparison of the three highest intense peptides of a protein (Hi3 method).
- Muntel et al. PMID: 24696501.